

Montelukast dosages: 10 mg, 5 mg, 4 mg
Montelukast packs: 30 pills, 60 pills, 90 pills, 120 pills, 180 pills, 270 pills, 360 pills

Buy montelukast 5 mg with mastercard
Paths of metabolism of chlorpromazine embrace hydroxylation and conjugation with glucuronic acid, N-oxidation, oxidation of a sulfur atom, and dealkylation. Potentially hazardous interactions with other medicine Anaesthetics: enhanced hypotensive impact. Analgesics: elevated risk of convulsions with tramadol; enhanced hypotensive and sedative effects with opioids; elevated risk of ventricular arrhythmias with methadone. Antibacterials: elevated danger of ventricular arrhythmias with moxifloxacin and telithromycin � keep away from with moxifloxacin. Antidepressants: increased stage of tricyclics, possibly elevated threat of ventricular arrhythmias and antimuscarinic side effects. Anticonvulsant: antagonises anticonvulsant impact; concentration of phenytoin possibly elevated or decreased; concentration of each medication reduced with phenobarbital. Antipsychotics: increased danger of ventricular arrhythmias with droperidol and pimozide � keep away from concomitant use; concentration of haloperidol presumably increased. Diabetes insipidus: 100 mg each 12 hours initially, reducing to 50 mg daily where attainable. Distribution within the blood is concentration-dependent, with 41�58% in erythrocytes and 10�20% in leucocytes; the rest is found in plasma, about 90% protein-bound, principally to lipoprotein. Ciclosporin is extensively metabolised within the liver and primarily excreted in faeces through the bile. Antibacterials: increased threat of myopathy with daptomycin � try to keep away from concomitant use. When ciclosporin and bosentan are coadministered, preliminary trough concentrations of bosentan are 30 occasions higher than regular. Colchicine: risk of myopathy or rhabdomyolysis; additionally elevated blood-ciclosporin concentrations and nephrotoxicity. Cytotoxics: elevated threat of neurotoxicity with doxorubicin; focus of epirubicin, everolimus and idarubicin elevated; decreased excretion of mitoxantrone; elevated toxicity with methotrexate; seizures have been reported in bone marrow transplant patients taking busulfan and cyclophosphamide; use crizotinib with caution; concentration of etoposide possibly elevated (increased danger of toxicity); attainable interplay with docetaxel. Lipid-lowering brokers: absorption reduced by colesevelam, increased risk of myopathy with statins (avoid concomitant use with simvastatin, max dose of atorvastatin ought to be 10 mg1); avoid with rosuvastatin; elevated threat of nephrotoxicity with fenofibrate; bezafibrate might improve creatinine and scale back ciclosporin ranges; focus of both medication could also be elevated with ezetimibe. Sirolimus: increased absorption of sirolimus � give sirolimus four hours after ciclosporin; sirolimus concentration increased; long term concomitant administration could also be related to deterioration in renal operate. Tacrolimus: increased ciclosporin concentration and toxicity � avoid concomitant use. Ursodeoxycholic acid: unpredictably increased absorption and raised ciclosporin ranges in some sufferers. Over 2�6 hours peripherally or 1 hour centrally Dilute 50 mg in 20�100 mL with sodium chloride 0. Cidofovir is eliminated mainly by renal excretion, both by glomerular filtration and tubular secretion. Use with probenecid might reduce the excretion of cidofovir to some extent by blocking tubular secretion, although 70�85% has still been reported to be excreted unchanged within the urine within 24 hours. Administer 2 hours before dialysis session to profit from peak focus with out having delayed clearance. Pharmacokinetics of cidofovir in renal insufficiency and in steady ambulatory peritoneal dialysis or high-flux haemodialysis. After oral absorption cilazapril is quickly metabolised in the liver to cilazaprilat, the bioavailability of which is about 60%. Peak plasma concentrations of cilazaprilat happen inside 2 hours of an oral dose of cilazapril. Antibacterials: focus increased by erythromycin � think about decreasing cilostazol dose. Antifungals: focus probably increased by ketoconazole � think about lowering cilostazol dose. Calcium-channel blockers: focus increased by diltiazem � think about decreasing cilostazol dose. Ulcer-healing drugs: concentration increased by omeprazole � consider decreasing cilostazol dose. The drug should be used with nice caution if administered to patients with a creatinine clearance <25 mL/min.
Buy montelukast 5mg with mastercard
The finding of a Y-bearing cell line suggests an increased danger for malignant tumor in the dysgenetic gonad, which ought to be removed. Any Case with a Visible Deletion, Duplication, or Unbalanced Translocation In this case, chromosome research must be done on each parents to rule out a rearrangement such as a pericentric inversion, a balanced translocation, or, in rare circumstances, an insertion, that would predispose to recurrence of the abnormality. If parental karyotypes are normal, as is the case in the majority of families, the recurrence threat is low. If a parental rearrangement is recognized, the theoretical threat for recurrence is elevated. For a number of the more common rearrangements, empiric danger figures are available in the literature. In the case of balanced translocation carriers, the earlier reproductive expertise of the couple have to be thought-about in counseling. Many, however not all, mother and father with microdeletions also categorical the phenotype to some degree. Recurrence threat for the genomic anomaly in these people is 50% for every subsequent being pregnant, but the severity of the phenotype that might ensue is commonly unpredictable, thus posing a challenge for genetic counseling. Identification of a brand new microdeletion or microduplication-neither beforehand found in the regular inhabitants with important frequency nor beforehand reported in affected sufferers with concordant phenotypes-is a challenge for interpretation. If de novo, the assumption of causality can only be based on gene content material, animal studies, and the concordance of the precise gene loss or duplication with a acknowledged phenotype. The literature and public databases should be looked for beforehand reported instances earlier than offering the family with vital data on prognosis and natural history. A human being is a diploid organism with two units of chromosomes, one set from every mother or father. Each pair of chromosomes may have comparable gene determinants located on the similar place on every chromosome pair. The pair of genes could additionally be referred to as alleles, or partners, which usually work collectively. Thus, excluding the genes of the X and Y chromosomes within the male and those of the mitochondria, each genetic determinant is present in two doses, one from each parent. However, for most genes on the X and for close to 100 genes on the autosomes, only a single copy of the gene is actively Microdeletion and Microduplication Syndromes Although microdeletion syndromes are chromosomal abnormalities, because the problem that produces the phenotype is genetic imbalance quite than genetic mutation and since the abnormality 880 Genetics, Genetic Counseling, and Prevention expressed (monoallelic expression). A main mutant gene is herein outlined as a genetic determinant that has modified in such a method that it can provide rise to an abnormal attribute. A mutant gene that causes an abnormal attribute when current in double dosage (or single dosage and not utilizing a normal associate, as for an X-linked mutant gene in the male) is referred to as "recessive. As more is discovered concerning the molecular biology of mutant genes, the distinction between dominant and recessive genes has blurred. Loss of one copy of the gene (haploinsufficiency) could cut back by half the gene product leading to useful alteration of development. Mutations may create proteins with both an elevated perform (gain of operate mutations) or a completely new perform (dominant adverse mutations) that may intrude with regular improvement as nicely. Interestingly, mutations causing haploinsufficiency in the gene may trigger one phenotype and people leading to acquire of function, a totally totally different one. The various forms of osteogenesis imperfecta are good examples of types of dominant mutations. One pair of regular genes is represented as dots on a homologous pair of chromosomes. The chance of inheritance of the mutant gene () is the same as the possibility of inheriting a selected chromosome of the pair: 50 percent. A single mutant gene is recessive () if it causes no evident abnormality, the perform being nicely covered by the normal associate gene (allele). The dad and mom are typically carriers, and their risk of having one other affected offspring is the prospect of receiving the mutant from one father or mother (50 percent) times the possibility from the opposite father or mother (50 percent), or 25 % for each offspring. An X-linked recessive shall be expressed within the male as a end result of he has no regular associate gene. His daughters, receiving the X, will all be carriers, and his sons, receiving the Y, will all be normal. Genetics, Genetic Counseling, and Prevention 881 by half; nonetheless, many biologic techniques are forgiving of quantitative decrease in gene function-hence the silence of recessive mutations when current in single copy (heterozygosity). Half of the normal amount of exercise of alpha iduronidase has no impact on the individual with the altered gene; nonetheless, the enzyme deficiency resulting from a double dose of the altered gene produces a extreme phenotype.
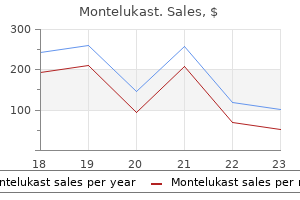
Buy montelukast australia
Letter: A new constant chromosomal abnormality in continual myelogenous leukaemia recognized by quinacrine fluorescence and Giemsa staining. Myelofibrosis in Philadelphia chromosome� negative myeloproliferative neoplasms is associated with aberrant karyotypes. Impact of bone marrow pathology on the clinical management of Philadelphia chromosome�negative myeloproliferative neoplasms. Trends in continual myeloid leukemia incidence and survival in the United States from 1975 to 2009. Detection of c-abl tyrosine kinase exercise in vitro permits direct comparison of normal and altered abl gene merchandise. The peripheral blood in continual granulocytic leukaemia: research of fifty untreated Philadelphia-positive instances. Favorable long-term follow-up outcomes over 6 years for response, survival, and safety with imatinib mesylate therapy in chronic-phase persistent myeloid leukemia after failure of interferon-alpha therapy. Chronic neutrophilic leukemia: novel mutations and their impact on medical apply. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical continual myeloid leukemia, and persistent myelomonocytic leukemia. Survival and prognosis amongst 1545 sufferers with contemporary polycythemia vera: a world study. Trends in the incidence of polycythemia vera among Olmsted County, Minnesota residents, 1935�1989. Aquagenic pruritus in polycythemia vera: characteristics and affect on quality of life in 441 patients. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Neutrophilic leukocytosis in superior stage polycythemia vera: hematopathologic options and prognostic implications. Acute leukemia in polycythemia vera: an evaluation of 1638 sufferers enrolled in a prospective observational study. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: a global examine of 797 patients. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976�1995. The syndrome of idiopathic myelofibrosis: a clinicopathologic review with emphasis on the prognostic variables predicting survival. Splenomegaly in 2,505 patients in a big college medical heart from 1913 to 1995. Survival and illness development in essential thrombocythemia are considerably influenced by accurate morphologic analysis: an international study. Morphologic and cytogenetic variations between post-polycythemic myelofibrosis and first myelofibrosis in fibrotic stage. High concordance in grading reticulin fibrosis and cellularity in patients with myeloproliferative neoplasms. Impact of allogeneic stem cell transplantation on survival of sufferers lower than 65 years of age with primary myelofibrosis. Exploratory evaluation of the effect of ruxolitinib on bone marrow morphology in sufferers with myelofibrosis. Disappearance of fibrosis in secondary myelofibrosis after ruxolitinib remedy: new endpoint to achieve Incidence, scientific options and consequence of important thrombocythaemia in a properly defined geographical space. Polycythemia vera and important thrombocythemia: 2015 replace on analysis, risk-stratification and administration. Incidence of myeloproliferative hypereosinophilic syndrome in the United States and an estimate of all hypereosinophilic syndrome incidence. Hypereosinophilic syndrome: a multicenter, retrospective evaluation of scientific traits and response to remedy.
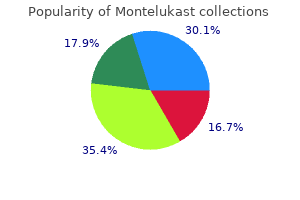
Montelukast 5mg mastercard
Females are symbolized by circles, males by squares, persons of unknown intercourse by diamonds. Affected people are represented by stable symbols, these unaffected, by open symbols. Marriages or matings are indicated by horizontal strains linking male and female symbols, with the male partner preferably to the left. Offspring are shown beneath the parental symbols, in start order from left to right, linked to the mating line by a vertical, and numbered (1, 2, 3, and so forth. The proband is indicated by an arrow with the letter P, the consultand by an arrow alone. Use of pedigrees A good household pedigree reveals the mode of inheritance of the illness and can be utilized to predict the genetic risk in a number of instances (see Chapter 13). These embrace: 1 the present being pregnant; 2 the chance for future offspring of these dad and mom (recurrence risk); three the danger of disease among offspring of shut family members; 4 the chance of adult disease, in cases of ailments of late onset. But, in distinction to fluid essences that ought to mix within the offspring in all proportions, Mendel confirmed that the instructions for contrasting characters segregate and recombine in easy mathematical proportions. Mendel postulated 4 new rules regarding unit inheritance, dominance, segregation and unbiased assortment that apply to most genes of all diploid organisms. Offspring of 4 genotypes are produced: RrFf, Rrff, rrFf and rrff and these are in the ratio 1: 1: 1: 1. These offspring even have phenotypes which might be all different: non-red with free earlobes, non-red with connected, purple with free, and purple with hooked up, respectively. The principle of unit inheritance Hereditary characters are determined by indivisible units of data (which we now name genes). The test-mating the mating described above, in which one associate is a double recessive homozygote (rrff), constitutes a test-mating, as his or her recessive alleles permit expression of all the alleles of their associate. However, because of dominance all have non-red hair and free earlobes, so the genotype of the heterozygous parent stays obscure. The principle of dominance Alleles occur in pairs in each individual, however the results of 1 allele could additionally be masked by these of a dominant companion allele. The principle of segregation During formation of the gametes the members of every pair of alleles separate, so that every gamete carries only one allele of every pair. Example the earlobes of some folks have an elongated attachment to the neck while others are free, a distinction we are able to consider for the purposes of this explanation to be determined by two alleles of the identical gene, f for connected, F for free. The youngsters constitute the first filial technology or F1 generation (irrespective of the symbol for the gene underneath consideration). Individuals with similar alleles are homozygotes; these with totally different alleles are heterozygotes. The second filial, or F2, era is composed of the grandchildren of the unique couple, resulting from mating of their offspring with partners of the identical genotype on this respect. In each case both parents are heterozygotes, so each produce F and f gametes in equal numbers. Due to the dominance of F over f, dominant homozygotes are phenotypically the same as heterozygotes, so there are three offspring with free earlobes to each one with hooked up. The phenotypic ratio 3: 1 is characteristic of the offspring of two heterozygotes. Matings between double heterozygotes the triumphant mathematical proof of Mendel legal guidelines was provided by matings between pairs of double heterozygotes. This allows us to predict the chances of producing: 1 a child with non-red hair and free earlobes (R-F-), as 9/16; 2 a baby with non-red hair and connected earlobes (R-ff), as 3/16; 3 a child with pink hair and free earlobes (rrF-), as 3/16; and four a baby with red hair and connected earlobes (rrff), as 1/16. The principle of impartial assortment Different genes control different phenotypic characters and the alleles of various genes re-assort independently of every other. Sex-relatedeffects the genetic specification of sexual differentiation is described in Chapter 43. The Y carries a small variety of genes concerned with growth and maturation of masculine options and likewise sections homologous with elements of the X. Y-linked traits (of which there are very few) are due to this fact confined to males, but X-linked can show a criss-cross sample from fathers to daughters, moms to sons down the generations. The most important aspect of sex-related inheritance issues X-linked recessive alleles, of which there are many.
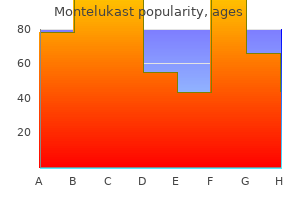
Order montelukast 5 mg amex
The mandibular teeth could turn into crowded, possibly requiring elimination of one or more. Todorov and colleagues developed a screening take a look at that establishes normal milestones for kids with achondroplasia as much as 2 years of age. There is an inclination towards late childhood weight problems, and females are more susceptible to have menorrhagia, fibroids, and enormous breasts. By 10 years of age, a big variety of affected youngsters have developed neurologic symptoms with claudication and elevated reflexes in their legs. Surgical leg lengthening is controversial because of the necessity for repeated surgeries, the lengthy time period that orthopedic appliances must be used, superficial infections, and the stretching of nonskeletal tissues. Megalocephaly, small foramen magnum, short cranial base with early sphenooccipital closure, low nasal bridge with outstanding forehead, mild midfacial hypoplasia with narrow nasal passages. Small cuboid-shaped vertebral our bodies with quick pedicles and progressive narrowing of lumbar interpedicular distance; lumbar lordosis, gentle thoracolumbar kyphosis with anterior beaking of first or second lumbar vertebra; small iliac wings with narrow larger sciatic notch; brief tubular bones, particularly humeri; metaphyseal flare with ball-and-socket arrangement of epiphysis to metaphysis; short trident hand, fingers being similar in size, with quick proximal and midphalanges; brief femoral neck; incomplete extension of elbow. Mild hypotonia; early motor progress is commonly sluggish, though eventual intelligence is often normal; relative glucose intolerance evident with an oral glucose tolerance take a look at. Therefore, ultrasound research of the mind should be performed if the fontanel measurement is particularly giant, the occipitofrontal circumference increases too rapidly, head circumference above the 95th percentile, or any signs of hydrocephalus develop. Respiratory problems secondary to a small chest, upper airway obstruction, and sleep-disordered respiratory are widespread. Indications for decompression embody lower limb hyper-reflexia, central apnea, and foramen magnum measurements below the imply for achondroplasia. Interestingly, virtually all circumstances demonstrate the identical single base pair substitution, presumably accounting for the consistency of the phenotype seen on this disorder. References Maroteaux P, Lamy M: Achondroplasia in man and animals, Clin Orthop 33:ninety one, 1964. Caffey J: Pediatric X-Ray Diagnosis, ed 5, Chicago, 1967, Year Book Medical Publishers. Oberklaid F, et al: Achondroplasia and hypochondroplasia, J Med Genet 16:a hundred and forty, 1979. Head development in achondroplasia: Use of ultrasound studies, Am J Med Genet 13:105, 1982. Brinkman G, et al: Cognitive skills in achondroplasia, Am J Med Genet 47:800, 1993. Cervicomedullary junction compression in infants with achondroplasia: When to carry out neurosurgical decompression, Am J Hum Genet 56:824, 1995. Georgoulis G, et al: Achondroplasia with synostosis of a quantity of sutures, Am J Med Genet 155:1969, 2011. A, Newborn toddler with achondroplasia, exhibiting macrocephaly, low nasal bridge, relatively small thoracic cage, shortness of humeri and femora (rhizomelia). Photograph of a 1-year-old woman displaying relative macrocephaly, small thoracic cage, and rhizomelic shortening. Note low nasal bridge, relative macrocephaly with distinguished forehead, and midface hypoplasia. A and B, Note in the newborn period, the "trident" hand with quick metacarpals and phalanges, caudal narrowing of spinal canal with brief pedicles, and small iliac wings with narrow higher sciatic notch. Macrocephaly, predominantly caused by a big brain, is a ordinary characteristic of individuals with achondroplasia. Hypochondroplasia has an incidence of approximately one twelfth that of achondroplasia and can be distinguished from it by the relative lack of craniofacial involvement and milder features in the arms and backbone. Patients with the N540K mutation have a more severe phenotype related to disproportionate quick stature, macrocephaly, and with radiologic proof of unchanged/narrow interpedicular distance and fibula longer than tibia. Relatively brief without rhizomelic, mesomelic, or acromelic predominance; brief tubular bones with delicate metaphyseal flare; brief, broad femoral necks; long distal fibulae, short distal ulnae, and long ulnar styloids; brachydactyly; bowing of legs; stubby arms and ft; mild limitation in elbow extension and supination. Anteroposterior shortening of lumbar pedicles on lateral view; spinal canal narrowing or unchanged caudally, with or without lumbar lordosis.
Bastard Cinnamon (Cassia Cinnamon). Montelukast.
- Dosing considerations for Cassia Cinnamon.
- Diabetes.
- What is Cassia Cinnamon?
- Are there any interactions with medications?
- Are there safety concerns?
- How does Cassia Cinnamon work?
- Loss of appetite, muscle and stomach spasms, bloating, intestinal gas, vomiting, diarrhea, common cold, impotence, bed wetting, menstrual complaints, chest pain, high blood pressure, kidney problems, cancer, and other conditions.
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96963
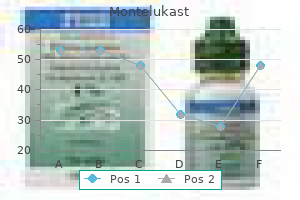
Generic montelukast 10 mg visa
No affected person has been in a place to walk or acquire expressive language other than babbling. Early remedy with beta-blockers might slow the development of cardiac deterioration. The instances with out mutations appeared to have longer survival and no cardiomyopathic manifestations. Vici syndrome is the first example of a human multisystem disorder associated with defective autophagy. Profound cervicoaxial hypotonia with hyperextended neck posture, flexed lower limbs, and clenched fists. Agenesis of the corpus callosum and septum pellucidum, cerebral atrophy with enlarged ventricles, hypoplasia of the cerebellar vermis and pons, gyral anomalies such as heterotopias or other types of non-lissencephalic cortical dysplasia, schizencephaly, delayed myelination, opercular hypoplasia, hypoplasia of optic nerves and chiasma. Postnatal onset microcephaly, ptosis, depressed nasal bridge, high palate, tented higher lip, delicate micrognathia. Congenital or acquired cataracts, hypopigmented retina and iris, hypoplasia of macula and optic disk, horizontal nystagmus, marked visual disturbance, lack of light reflex, inconsistent visible fixation and tracking, gradual or absent visual evoked potentials, photophobia. Hypopigmentation of the skin, ranging from lighter complexion to full albinism. Broad spectrum of defects starting from a mixed immunodeficiency to a nearly regular immunity. Miyata R, et al: Sibling instances of Vici syndrome: Sleep abnormalities and complications of renal tubular acidosis, Am J Med Genet A 143:189, 2007. McClelland V, et al: Vici syndrome associated with sensorineural hearing loss and proof of neuromuscular involvement on muscle biopsy, Am J Med Genet A 152A:741, 2007. Finocchi A, et al: Immunodeficiency in Vici syndrome: A heterogeneous phenotype, Am J Med Genet A 158A:434, 2012. Said E, et al: Vici syndrome-A rapidly progressive neurodegenerative dysfunction with hypopigmentation, immunodeficiency and myopathic modifications on muscle biopsy, Am J Med Genet A 158A:440, 2012. There is marked generalized hypopigmentation relative to the ethnic background (E, F). Coarsening of facial options with full lips and macroglossia is noted in some older children (G). The cardiac anomalies are normally not as severe as these with bilateral right-sidedness. The advanced cardiac anomalies, normally giving rise to cyanosis and early cardiac failure, are the main explanation for early dying. The chance of gastrointestinal issues should even be considered, especially as associated to the aberrant mesenteric attachments. Among other variations, the spleen dramatically displays the variant laterality in the two. Left-right axis malformations are normally isolated but can occur as one function of a multiple malformation syndrome, the most typical of which is immotile cilia syndrome. As a result of defective cilia and flagella, persistent respiratory tract infections occur commonly, and infertility in males, chronic ear infections, and decreased or absent odor occur variably. The cilia are functionally abnormal and, on electron microscopy, have absent or irregular dynein arms connecting the nine pairs of microtubules. A subgroup of the immotile cilia Laterality Sequences 797 associated to the asplenia. Tests to detect asplenia embrace analysis of pink blood cells for HowellJolly bodies and Heinz bodies. As such, although often sporadic, autosomal dominant, autosomal recessive, and X-linked recessive inheritance have all been documented. At present, only 10% of circumstances are attributable to mutations of recognized genes, although additional candidate genes have emerged from studies of left-right axis improvement in vertebrates. In the mouse, motile embryonic cilia generate directional move of extraembryonic fluid surrounding the node positioned on the tip of the embryo within the midline. This circulate concentrates left-right determinants to one side of the node activating uneven gene expression on the node and past. In the chick, activin on the best side of the primitive streak represses expression of the gene sonic hedgehog (Shh). The remaining expression of Shh on the left induces nodal on the left, leading to the conventional looping of the guts tube to the right. U 798 U Miscellaneous Sequences McGrath J, Brueckner M: Cilia are on the heart of vertebrate left-right asymmetry, Curr Opinion Genet Devel thirteen:385, 2003.
5 mg montelukast with mastercard
Deafness, probably the most serious characteristic, is sensorineural, congenital, and normally nonprogressive. It may be unilateral or bilateral and varies from slight to profound, though often the latter. The defect seems to be in the organ of Corti, with atrophic modifications in the spiral ganglion and nerve. Lateral displacement of inner canthi with brief palpebral fissures and lateral lacrimal dystopia; broad and high nasal bridge with hypoplastic alae nasi; medial flare of bushy eyebrows, which may meet in midline; hypochromic iridis; partial albinism manifested by hypopigmented ocular fundus and white eyelashes, eyebrows, and forelock. Deafness, aplasia of the posterior semicircular canal, untimely graying, broad mandible. Patent metopic suture, strabismus, rounded tip of nostril, full lips with accentuated "cupid bow" to higher lip, easy philtrum, cleft lip and palate, anisocoria, cardiac anomaly (ventricular septal defect), imperforate anus, Sprengel anomaly, supernumerary vertebrae and ribs, neural tube closure defect, scoliosis, multicystic dysplastic kidney, absence of vagina and adnexa uteri. Features of kind I with the addition of higher limb defects, together with hypoplasia of muscle tissue, flexion contractures, carpal bone fusion, and syndactyly. Pingault V, et al: Review and update of mutations inflicting Waardenburg syndrome, Hum Mutat 31:391, 2010. A and B, Note the lateral displacement of the medial canthi, the broad nasal bridge with hypoplastic ala nasi, medial eyebrow flare, and hypochromic iridis. A, Note the lateral displacement of the inner canthi (telecanthus) with brief palpebral fissures, anisochromia of irides, bushy eyebrows with medial flare, broad and high nasal bridge, a rounded tip of the nostril, and a smooth philtrum. B and C, A white hair forelock and skin changes over her leg are reflective of patchy hypopigmentation. In the Nineteen Forties, Franceschetti and Klein made intensive stories on this situation and referred to as it mandibulofacial dysostosis. The development of the facial bones during infancy and childhood leads to some beauty enchancment which could be enhanced by plastic surgery. Franceschetti A, Klein D: the Mandibulofacial Dysostosis: A New Hereditary Syndrome, Copenhagen, 1949, E. Peterson-Falzone S, Pruzansky S: Cleft palate and congenital palatopharyngeal incompetency in mandibulofacial dysostosis, Cleft Palate J 13:354, 1976. The Treacher Collins Syndrome Collaborative Group: Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome, Nat Genet 12:one hundred thirty, 1996. As the good majority of these patients are of normal intelligence, the early recognition of deafness and its correction with hearing aids or surgery are of great significance for improvement. Writzl K, et al: Genital anomalies in a affected person with Treacher Collins syndrome, Am J Med Genet 146:2169, 2008. Note the hair extending onto the lateral cheek, downslanting palpebral fissures, malar hypoplasia, malformed ears, and micrognathia. E and F, Note evidence of a cleft of the zygomatic bone and the coloboma of each upper and lower lids. In 2012, Lines and colleagues recognized the causative gene and extra utterly described the entire pattern of malformation, which is clearly distinct from Treacher Collins syndrome. Metopic ridge, malar hypoplasia, micrognathia, upslanting or downslanting palpebral fissures, microtia, preauricular tags, stenosis/atresia of exterior auditory canals, conductive hearing loss, cleft palate, choanal atresia. Wieczorek D, et al: Microcephaly, microtia, preauricular tags, choanal atresia and developmental delay: A mandibulofacial dysostosis distinct from Treacher Collins syndrome, Am J Med Genet A 149A:837, 2009. Affected child as a new child (A and B), at age three months (C and D), at age 10 months (E), at age 3 years (F), and at age 7 years (G). Note the prominent nasal bridge, dysplastic ears, malar hypoplasia, microretrognathia, and hypoplastic thumb (H). Short depressed nostril with flat nasal bridge and anteverted nares; look of enormous eyes; flat midface; distinguished, protruding higher incisors; thick lips. Sensorineural or blended loss, primarily affecting high frequencies and normally progressive. Calvarial thickening; absent frontal sinuses; falx, tentorial, and meningeal calcifications; spondyloepiphyseal abnormalities, together with delicate platyspondyly, barely small and irregular distal femoral and proximal tibial epiphyses, outward bowing of radius and ulna, and wide tufts of distal phalanges. Hearing loss has been famous in early childhood and sometimes progresses to moderate or extreme by adulthood.

Generic montelukast 4 mg with visa
Many survivors are blind and deaf and interact only minimally with their surroundings. In one case an interstitial deletion at 3p25p26, thought to be the smallest 3p deletion related to the characteristic phenotype, was reported. Microcephaly with flat occiput, synophrys, epicanthal folds, ptosis, quick palpebral fissures, prominent nasal bridge, small nostril with anteverted nares, lengthy philtrum, malformed ears, micrognathia, downturned corners of mouth. References Verjaal M, De Nef J: A patient with a partial deletion of the brief arm of chromosome 3, Am J Dis Child 132:43, 1978. Schwyzer U, et al: Terminal deletion of the short arm of chromosome 3, del(3pter-p25): A recognizable syndrome, Helv Paediatr Acta forty two:309, 1987. Nienhaus H, et al: Infant with del(3)(p25-pter): Karyotypephenotype correlation and evaluate of previously reported cases, Am J Med Genet 44:573, 1992. Hirschhorn and colleagues carried out chromosome banding studies in 1973 that related duplication of the 3q21qter region with a definite phenotype that Francke and Opitz subsequently emphasised may be clinically distinguished from Brachmann�de Lange syndrome. For survivors, intellectual incapacity, progress retardation, and pulmonary infections are the rule. Seventy-five percent of instances come up from a segregation of a parental rearrangement. Abnormal head shape, frequently because of craniosynostosis; hypertrichosis and synophrys; upslanting palpebral fissures; broad nasal root; anteverted nares; prominent maxilla; lengthy philtrum; downturned corners of mouth; high-arched palate; cleft palate; micrognathia; malformed ears; quick, webbed neck. Fifth finger clinodactyly, hypoplastic nails, simian crease, talipes equinovarus arch dermal ridge sample or digital pattern with low ridge counts. Cardiac defects, chest deformities, renal or urinary tract anomalies, genital anomalies (primarily cryptorchidism), umbilical hernia. Hirschhorn K, et al: Precise identification of varied chromosomal abnormalities, Ann Hum Genet 36:3875, 1973. Steinbach P, et al: the dup(3q) syndrome: Report of eight cases and review of the literature, Am J Med Genet 10:159, 1981. Aqua M, et al: Duplication 3q syndrome: Molecular delineation of the important region, Am J Med Genet fifty five:33, 1995. Autoradiographic labeling research revealed that the deficit chromosome was a number 4 somewhat than a quantity 5. Expressive language is extremely limited, though 6% have been capable of pronounce simple sentences. Major feeding difficulties, usually requiring gastrostomy, are a major downside in infancy. The scientific phenotype is set by 4p deletions that embrace the terminal 4p16. A newly outlined important area inside an interval of 300�600 kb between the loci D4S3327 and D4S98-D4S168 has been discovered. The size of the deletion, the occurrence of complicated chromosome anomalies, and the severity of seizures are prognostic components. Eighty-seven percent of instances characterize de novo deletions, whereas in 13% of instances, one of the mother and father is a balanced translocation provider. In these circumstances in which the dysfunction is suspected clinically however commonplace chromosome research are normal, molecular approaches can be utilized. Microcephaly, "Greek warrior helmet" appearance of nose, high brow, prominent glabella, extremely arched eyebrows, strabismus, eye or optic nerve defects, higher lid/iris/chorioretinal coloboma, ocular hypertelorism, epicanthal folds, cleft lip and/or palate, downturned "fishlike" mouth, quick higher lip and philtrum, micrognathia, posterior midline scalp defects, cranial asymmetry, preauricular tag or pit. Hypoplastic dermal ridges, low dermal ridge count, simian creases, talipes equinovarus, hyperconvex fingernails. Hypospadias, cryptorchidism, clitoral hypoplasia, sacral dimple or sinus; cardiac anomalies, together with atrial septal defect, pulmonary stenosis, ventricular septal defect, and patent ductus arteriosis; scoliosis. Wolf U, Reinwein H: Klinische und cytogenetische Differentialdiagnose der Defizienzen an den kurzen Armen der B-Chromosomen, Z Kinderheilkd 98:235, 1967. Fagan-Bagric K, et al: A sensible application of fluorescent in situ hybridization to the Wolf-Hirschhorn syndrome, Pediatrics ninety three:826, 1994. Sharathkumar A, et al: Malignant hematological problems in kids with Wolf-Hirschhorn syndrome, Am J Med Genet 119:164, 2003.
References
- Enriquez-Sarano M, Tajik AJ, Bailey KR, Seward JB. Color flow imaging compared with quantitative Doppler assessment of severity of mitral regurgitation: influence of eccentricity of jet and mechanism of regurgitation. J Am Coll Cardiol 1993; 21(5):1211-1219.
- Chan BL,Witt R, Charrow AP, et al. Mirror therapy for phantom limb pain. N Engl J Med 2007;357: 2206-2207.
- Semelka RC, Ascher SM, Reinhold C: MRI of the Abdomen and Pelvis. Wiley-Liss, NewYork, 1997.
- Argenziano M, Katz M, Bonatti J, et al. Results of the prospective multicenter trial of robotically assisted totally endoscopic coronary artery bypass grafting. Ann Thorac Surg. 2006;81: 1666-74.
- Catalona WJ, Partin AW, Slawin KM, et al: Use of the percentage of free prostate-specific antigen to enhance differentiation of prostate cancer from benign prostatic disease: a prospective multicenter clinical trial, J Am Med Assoc 279:1542n1547, 1998.
- Barash PG, Glanz S, Katz JD, et al: Ventricular function in children during halothane anesthesia: An echocardiographic evaluation, Anesthesiology 49:79-85, 1978.
- Norlen LJ, Blaivas JG: Unsuspected proximal urethral obstruction in young and middle-aged men, J Urol 135:972n976, 1986.


